Dear Napoleão, Thank you for updating everyone on your efforts, and also acknowledging the advice.
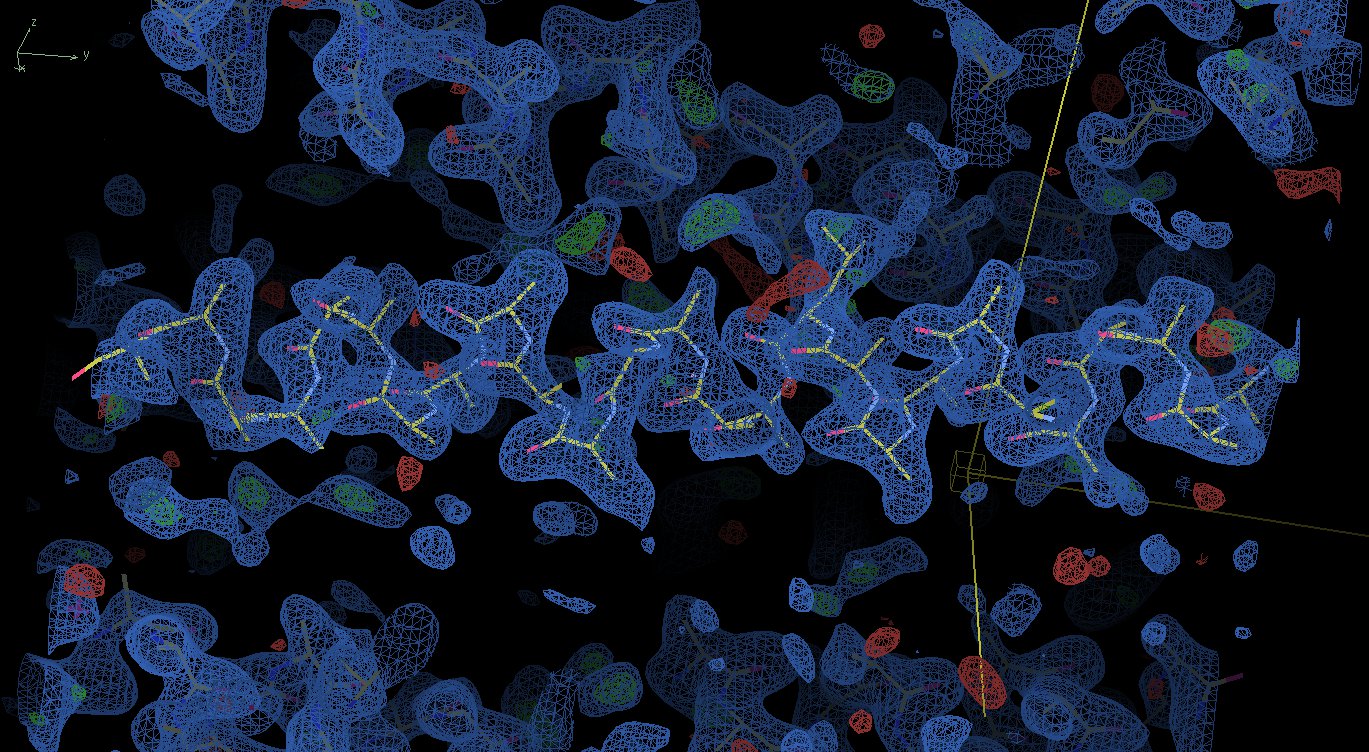
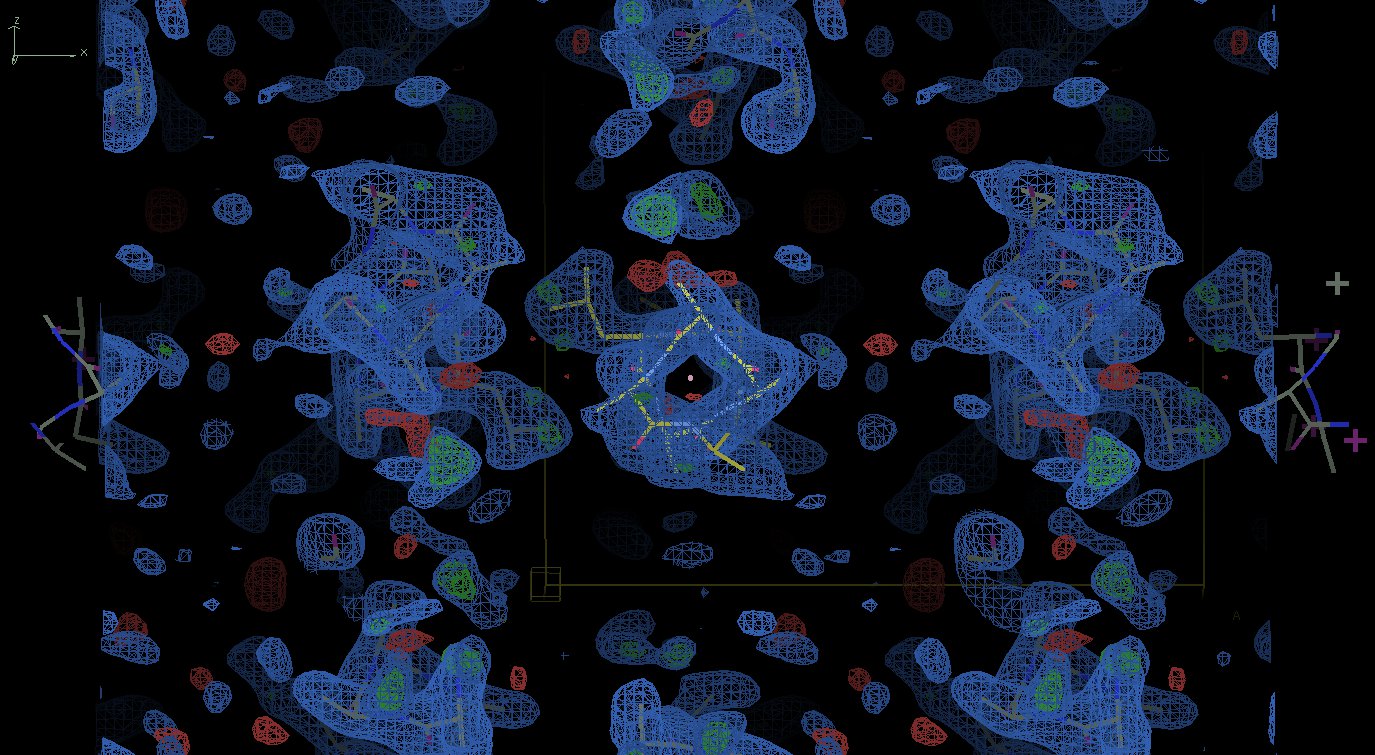
I wanted to respond to your question regarding maps. I know that many people who try to figure out whether or not their MR solution is the right one would ask the same question. So first of all if you wonder why you actually get very decently looking maps the answer is a classical one: because 'the phases are more important than amplitudes'. The appearance of your map is defined by your model phases, and hence a good match between the model and the map /may not/ be taken as a sign of a correct solution. Once again: /never ever!/ On the contrary, at least in your coil1.jpg image I clearly see that the density exactly follows the model which is almost entirely poly-Ala. Unless your protein is really poly-Ala this should be alarming. If you had a correct solution they you would hope to see the (difference)density for at least some missing side chains. And a second point. Unless your model contains the complete chain (which is rarely the case, especially for the coiled coils, as discussed already) a sign of the correct solution would be the appearance of extra density near the N- and/or C-terminus of the model. If it is not there, it is almost certainly not a solution. And you should not be worried about the R-factors being very high at this stage. If the solution is correct then you should see at least some extra features in the map. Kind regards, Sergei
Thank you all for the replies. Sorry for taking so long to reply, I was actually trying some of your interesting ideas (and I'm still trying). I tried using the low resolution data sets for the molecular replacement (thanks to Yuriy Patskovsky), I also improved and increased my coiled coil database and employed it in many approaches using EPRM (interesting program I was not aware of), which I found to produce lots of data, hopefully addressing at some extent the helixes bent (thanks to Bernhard Rupp). I also tried some more tweaking in Phaser, although not sure if did it properly (thanks to Randy Read). There is no twinning as far as I can tell (thanks to Ed Pozharski for the tip). Using a data set with enough completeness (360 degrees @ Brookhaven) and processing in P1 did not help me because in this space group there is most likely 2-3 helixes in the asymmetric unit, which complicates the problem (and it takes a lot of time for Phaser to run). Automated approaches also did not yield a better result (as far as I can tell). I'm convinced that the space group is C2221, but I may be wrong. Thanks to Sergei Strelkov for the numerous useful suggestions on how to approach the problem. One of the big issues for me is to discriminate between a lot of similarly good density maps. For example: http://www.fullonline.org/coils/coil1.jpg http://www.fullonline.org/coils/coil2.jpg I have hundreds of solutions like these and I think they are all wrong. I couldn't manage to run Arcimboldo, could not find a tutorial on it either. It was highly recommended here (and elsewhere), so I'm definitely willing to give it a try (thanks Isabel Uson). You guys opened my eyes about a series of issues that I should learn about and approach, I'm most thankful for that. Best regards, Napo
{kind=link}
{kind=link}
-- Prof. Sergei V. Strelkov Laboratory for Biocrystallography Department of Pharmaceutical Sciences, Katholieke Universiteit Leuven O&N2, Campus Gasthuisberg, Herestraat 49 bus 822, 3000 Leuven, Belgium Work phone: +32 16 330845 Fax: +32 16 323469 OR +32 16 323460 Mobile: +32 486 294132 Lab pages: http://pharm.kuleuven.be/anafar