Identité Message:<[EMAIL PROTECTED]>
Identité Message:<[EMAIL PROTECTED]>
X-Sender: [EMAIL PROTECTED]
X-Mailer: QUALCOMM Windows Eudora Pro Version 4.0.1
Date: Thu, 31 Dec 1998 19:02:22 +0100
To: [EMAIL PROTECTED]
From: Armel Le Bail <[EMAIL PROTECTED]>
Subject: Re: Chemical analysis of the year
References: <>
Mime-Version: 1.0
Content-Type: text/plain; charset="us-ascii"
Lachlan wrote :
>If not too rude a question - how good were
>the structures derived from the "false"
>chemical information (taken on good
>faith as being correct)?
>Fit, bond lengths and angles? Only
>a cursory comment is made about the
>alternatives solutions that were submitted.
I can answer as a participant. First of all, the
Dupont Challenge was not a challenge
because of structure complexity (13 independent
atoms is not really complex). The challenge was
that the material was ill-crystallized, showing
strong anisotropy of reflection broadening.
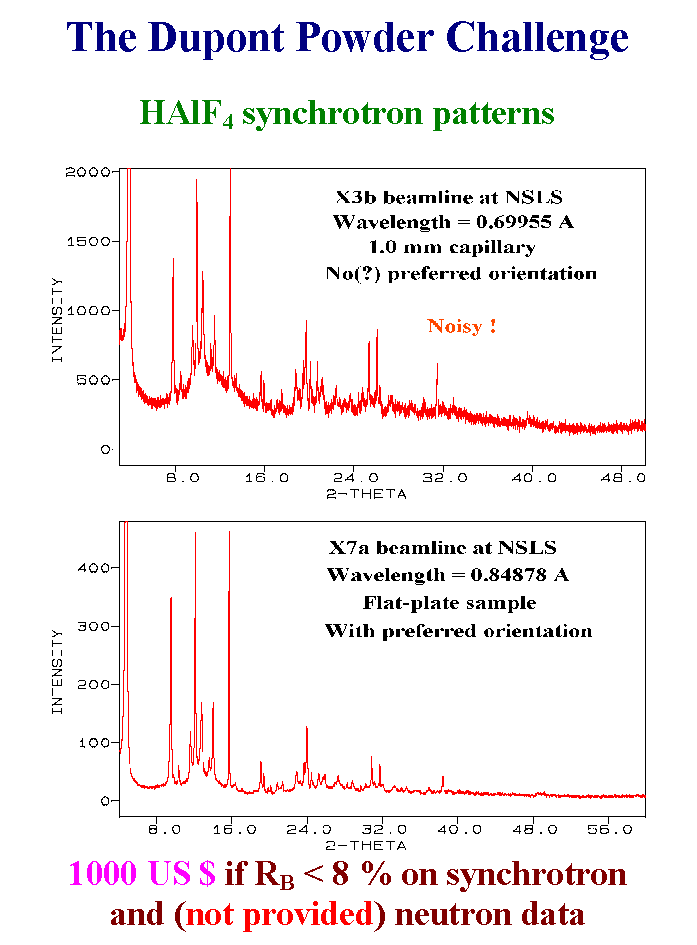
The challenge was that the organizers expected
the participant to obtain RB as low as 8% for
both neutron and X-ray data, while providing
only low quality X-ray synchrotron data (one noisy
pattern and one highly affected by preferred
orientation). Apart from the big formula mistake,
this was a real challenge !
Anyway, I had no special difficulty in providing
the Al3F10 octahedra framework, plus a possible
intersheet atom. Interatomic distances were acceptable,
but RB was near of 20% for both synchrotron pattern.
At a second step of the Dupont Challenge, when the
organizers realized that it was impossible, the neutron
data were provided. I was surprised by the low quality
pattern : noisy with a very high background indicating
that there could be more H atoms than expected from
a HAlF4 formula. I could proposed something that could
have been (H5O2)Al3F10, but no proposition with
HAlF4 formula could correspond to RB lower than 12%.
Those results were shown at ACA'1997, St Louis.
No news since that, although the organizers agreed
that new data were necessary.
>While single crystal has the reputation of being
>able to correct the synthetic chemist - what does
>this imply for solving structures from Powders?
This is true also for powder diffraction, in a lesser
extent of course, because Fourier syntheses are
not as clear as from single crystal data. The winners
apparently found the result in this way. But, if I have
well understood, they have used other diffraction data
than those of the original challenge (which I have never
seen...).
>Jumping to conclusions with near zilch hard
>information. The solution/refinement sequence is
>not described but the webpage mentions co-ordinates
>being refined using GSAS. GSAS also
>has convenient fourier electron density map
>generation and viewing. What is the corellation in
>the "goodness" of the "submitted" structure vs the
>refinement software's ability to conveniently generate
>and view fourier maps ? Especially with poor quality
>XRD data. Wouldn't refining without the routine aid
>of Fourier maps be like flying blind to some extent?
>
>Or is examining electron density space considered
>irrelevant in modern structure solution/refinement
>from powder data?
That question is curious. Of course, every serious
crystallographer examine electron density. The fact
is that it is not always clear. I personally use "|Fobs|"
from FULLPROF, examined with SHELXL. Have a look
at the original data, and you will understand why the
Fourier synthesis could not be clear :
http://www.cristal.org/iniref/aca97/t16.gif
I would like to see now the new data used for obtaining
the current proposition, a bit late for participating ;-).
However, maybe I am wrong and the challenge was
really won exclusively from the original data ?
Armel Le Bail - Universite du Maine, Laboratoire des Fluorures,
CNRS ESA 6010, Av. O. Messiaen, 72085 Le Mans Cedex 9, France
http://www.cristal.org/
{kind=link}