Dear Justin,
I'm pretty sure my .gro file is valid, here is a direct copy and paste of the
format of my .gro in the beginning:
star polymer
201
1star O4 1 0.000 0.000 0.000
1star O1 2 0.430 0.000 0.000
1star O1 3 0.860 0.000 0.000
1star O1 4 1.290 0.000 0.000
1star O1 5 1.720 0.000 0.000
1star O1 6 2.150 0.000 0.000
1star O1 7 2.580 0.000 0.000
1star O1 8 3.010 0.000 0.000
1star O1 9 3.440 0.000 0.000
1star O1 10 3.870 0.000 0.000
1star O1 11 4.300 0.000 0.000
1star O1 12 4.730 0.000 0.000
1star O1 13 5.160 0.000 0.000
1star O1 14 5.590 0.000 0.000
1star O1 15 6.020 0.000 0.000
1star O1 16 6.450 0.000 0.000
1star O1 17 6.880 0.000 0.000
1star O1 18 7.310 0.000 0.000
1star O1 19 7.740 0.000 0.000
1star O1 20 8.170 0.000 0.000
...
and for the output .gro after editconf, here is what it shows (I don't know why
the formats are kind of shifted, )
star polymer
201
1star O4 1 11.921 11.921 16.195
1star O1 2 12.351 11.921 16.195
1star O1 3 12.781 11.921 16.195
1star O1 4 13.211 11.921 16.195
1star O1 5 13.641 11.921 16.195
1star O1 6 14.071 11.921 16.195
1star O1 7 14.501 11.921 16.195
1star O1 8 14.931 11.921 16.195
1star O1 9 15.361 11.921 16.195
1star O1 10 15.791 11.921 16.195
1star O1 11 16.221 11.921 16.195
1star O1 12 16.651 11.921 16.195
1star O1 13 17.081 11.921 16.195
1star O1 14 17.511 11.921 16.195
1star O1 15 17.941 11.921 16.195
...
Another reason why I think my .gro is valid is that when I editconf, it says:
Read 201 atoms …blah blah blah.. , as it should I'm assuming if the .gro is
valid?
Please let me know, thank you.
Xu Dong Huang
Chemical & Biochemical Engineering
Rutgers School of Engineering
xudo...@eden.rutgers.edu
On Dec 31, 2012, at 5:40 PM, Xu Dong Huang <xudo...@eden.rutgers.edu> wrote:
> Dear Justin,
>
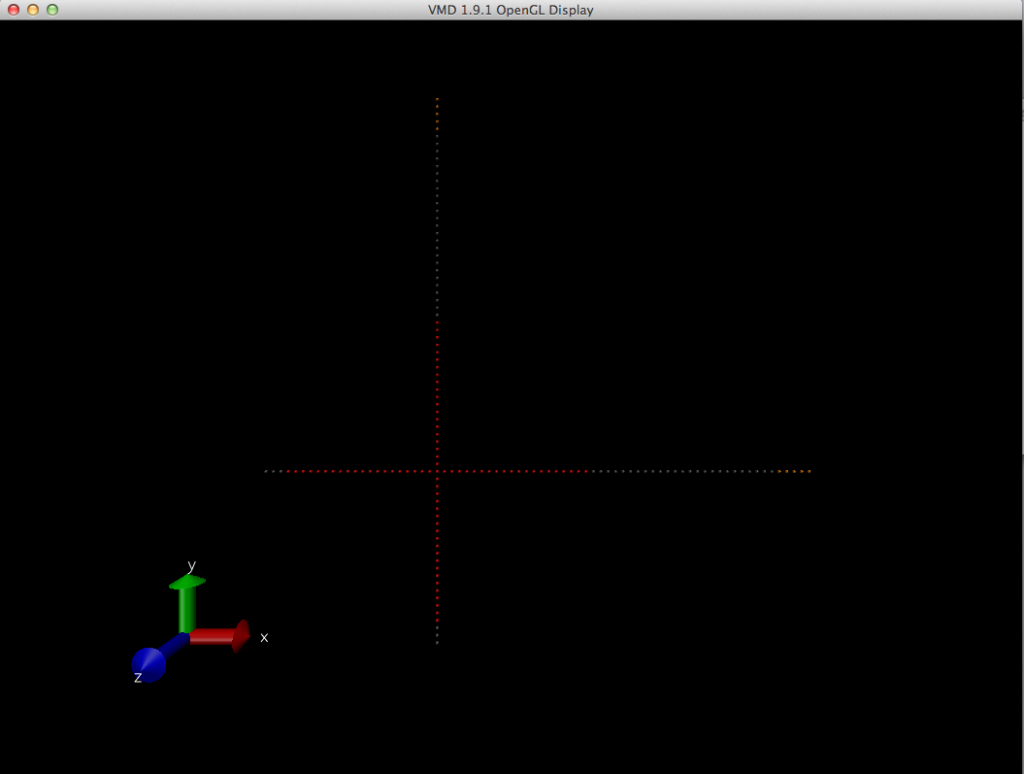
> (here is the picture of the molecule after editconf, note the negative x and
> negative y axis is missing the rest of the beads that is suppose to be like
> the positive x and positive y)
> http://i1284.photobucket.com/albums/a571/X_huang1/ScreenShot2012-12-31at53618PM_zpse5f421f1.png
>
> *Even though according to the .gro coordinates, those beads of 175 and up are
> jumped, i don't see the relocated beads anyhow in this VMD viewing. I'm so
> confused of what is happening.
>
>
> Xu Dong Huang
> Chemical & Biochemical Engineering
> Rutgers School of Engineering
> xudo...@eden.rutgers.edu
>
> On Dec 31, 2012, at 5:31 PM, Justin Lemkul <jalem...@vt.edu> wrote:
>
>>
>>
>> On 12/31/12 4:53 PM, Xu Dong Huang wrote:
>>> Dear advanced gromacs users,
>>>
>>> I have created an arbitrary molecule .gro file containing coordinates of my
>>> martini-beads. basically, there are 50 beads sitting on each axis of (x,y,
>>> -x and -y) forming like a cross. (Each bead with distance 0.43). However,
>>> when I try to use editconf on my .gro file to generate a box (editconf -f
>>> star.gro -o star_box.gro -c -d 1.0 -bt cubic) , it gives me back a new .gro
>>> file with a bad molecule. In VMD viewing, I see that the negative axis part
>>> of my molecules are cut off. Closer inspection shows the following for a
>>> particular segment of the molecule coordinates:
>>>
>>> **original .gro coordinates:
>>> 1star O1 170 0.000 -8.170 0.000
>>> 1star O1 171 0.000 -8.600 0.000
>>> 2star O2 172 0.000 -9.030 0.000
>>> 2star O2 173 0.000 -9.460 0.000
>>> 2star O2 174 0.000 -9.890 0.000
>>> 2star O2 175 0.000 -10.320 0.000
>>> 2star O2 176 0.000 -10.750 0.000
>>> 2star O2 177 0.000 -11.180 0.000
>>> 2star O2 178 0.000 -11.610 0.000
>>> 2star O2 179 0.000 -12.040 0.000
>>> 2star O2 180 0.000 -12.470 0.000
>>> 2star O2 181 0.000 -12.900 0.000
>>> 2star O2 182 0.000 -13.330 0.000
>>> 2star O2 183 0.000 -13.760 0.000
>>> *new .gro coordinates:
>>>
>>> 1star O1 169 12.421 4.681 16.695
>>> 1star O1 170 12.421 4.251 16.695
>>> 1star O1 171 12.421 3.821 16.695
>>> 2star O2 172 12.421 3.391 16.695
>>> 2star O2 173 12.421 2.961 16.695
>>> 2star O2 174 12.421 2.531 16.695
>>> 2star O2 175 12.421 22.741 16.695
>>> 2star O2 176 12.421 23.171 16.695
>>> 2star O2 177 12.421 23.601 16.695
>>> 2star O2 178 12.421 24.031 16.695
>>> 2star O2 179 12.421 24.461 16.695
>>> 2star O2 180 12.421 24.891 16.695
>>> 2star O2 181 12.421 25.321 16.695
>>> 2star O2 182 12.421 25.751 16.695
>>> 2star O2 183 12.421 26.181 16.695
>>>
>>> Notice the jump on atom number 174 and 175, why is there a sudden
>>> coordinate jump?
>>> All i can see in VMD is my negative axis part of the molecule beads are cut
>>> off.
>>>
>>
>> editconf builds boxes from the coordinate origin so that there are never
>> negative coordinates. It should be able to deal with those coordinates,
>> equally shifted, which it appears they were. Can you provide images of
>> what's going on? I don't clearly understand what's wrong. What do you mean
>> by "cut off?" Nonexistent? Incorrectly placed?
>>
>> -Justin
>>
>> --
>> ========================================
>>
>> Justin A. Lemkul, Ph.D.
>> Research Scientist
>> Department of Biochemistry
>> Virginia Tech
>> Blacksburg, VA
>> jalemkul[at]vt.edu | (540) 231-9080
>> http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin
>>
>> ========================================
>> --
>> gmx-users mailing list gmx-users@gromacs.org
>> http://lists.gromacs.org/mailman/listinfo/gmx-users
>> * Please search the archive at
>> http://www.gromacs.org/Support/Mailing_Lists/Search before posting!
>> * Please don't post (un)subscribe requests to the list. Use the www
>> interface or send it to gmx-users-requ...@gromacs.org.
>> * Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
>
> --
> gmx-users mailing list gmx-users@gromacs.org
> http://lists.gromacs.org/mailman/listinfo/gmx-users
> * Please search the archive at
> http://www.gromacs.org/Support/Mailing_Lists/Search before posting!
> * Please don't post (un)subscribe requests to the list. Use the
> www interface or send it to gmx-users-requ...@gromacs.org.
> * Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
--
gmx-users mailing list gmx-users@gromacs.org
http://lists.gromacs.org/mailman/listinfo/gmx-users
* Please search the archive at
http://www.gromacs.org/Support/Mailing_Lists/Search before posting!
* Please don't post (un)subscribe requests to the list. Use the
www interface or send it to gmx-users-requ...@gromacs.org.
* Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
{kind=link}