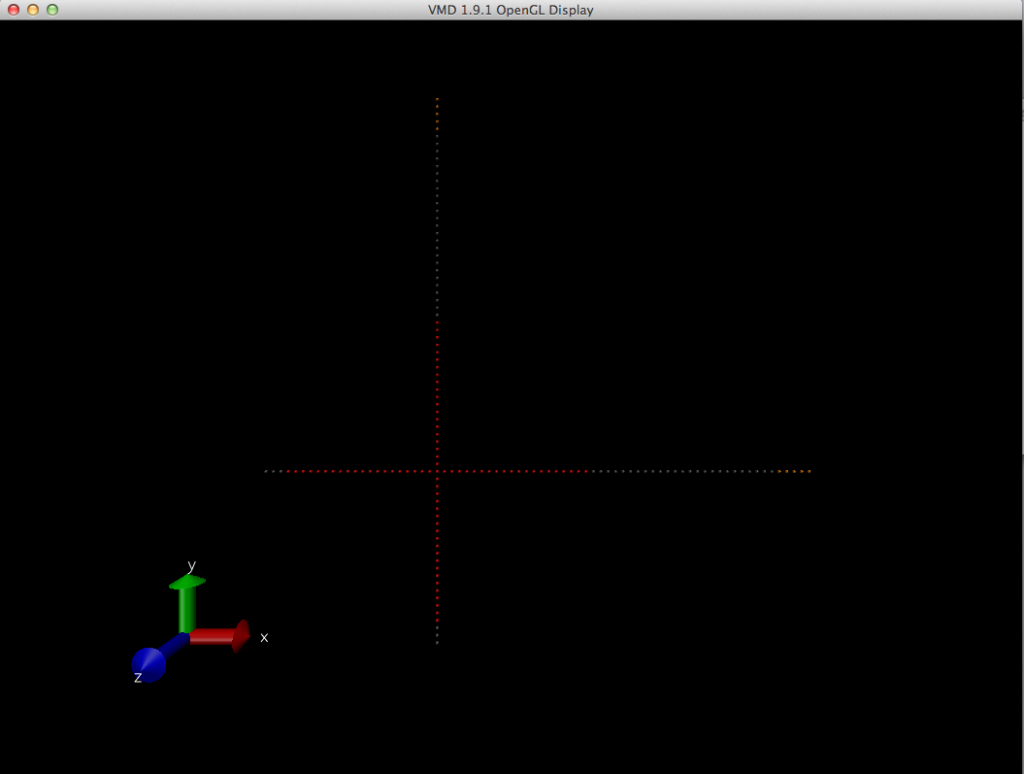
Dear Justin, (here is the picture of the molecule after editconf, note the negative x and negative y axis is missing the rest of the beads that is suppose to be like the positive x and positive y) http://i1284.photobucket.com/albums/a571/X_huang1/ScreenShot2012-12-31at53618PM_zpse5f421f1.png
{kind=link}
*Even though according to the .gro coordinates, those beads of 175 and up are jumped, i don't see the relocated beads anyhow in this VMD viewing. I'm so confused of what is happening. Xu Dong Huang Chemical & Biochemical Engineering Rutgers School of Engineering xudo...@eden.rutgers.edu On Dec 31, 2012, at 5:31 PM, Justin Lemkul <jalem...@vt.edu> wrote: > > > On 12/31/12 4:53 PM, Xu Dong Huang wrote: >> Dear advanced gromacs users, >> >> I have created an arbitrary molecule .gro file containing coordinates of my >> martini-beads. basically, there are 50 beads sitting on each axis of (x,y, >> -x and -y) forming like a cross. (Each bead with distance 0.43). However, >> when I try to use editconf on my .gro file to generate a box (editconf -f >> star.gro -o star_box.gro -c -d 1.0 -bt cubic) , it gives me back a new .gro >> file with a bad molecule. In VMD viewing, I see that the negative axis part >> of my molecules are cut off. Closer inspection shows the following for a >> particular segment of the molecule coordinates: >> >> **original .gro coordinates: >> 1star O1 170 0.000 -8.170 0.000 >> 1star O1 171 0.000 -8.600 0.000 >> 2star O2 172 0.000 -9.030 0.000 >> 2star O2 173 0.000 -9.460 0.000 >> 2star O2 174 0.000 -9.890 0.000 >> 2star O2 175 0.000 -10.320 0.000 >> 2star O2 176 0.000 -10.750 0.000 >> 2star O2 177 0.000 -11.180 0.000 >> 2star O2 178 0.000 -11.610 0.000 >> 2star O2 179 0.000 -12.040 0.000 >> 2star O2 180 0.000 -12.470 0.000 >> 2star O2 181 0.000 -12.900 0.000 >> 2star O2 182 0.000 -13.330 0.000 >> 2star O2 183 0.000 -13.760 0.000 >> *new .gro coordinates: >> >> 1star O1 169 12.421 4.681 16.695 >> 1star O1 170 12.421 4.251 16.695 >> 1star O1 171 12.421 3.821 16.695 >> 2star O2 172 12.421 3.391 16.695 >> 2star O2 173 12.421 2.961 16.695 >> 2star O2 174 12.421 2.531 16.695 >> 2star O2 175 12.421 22.741 16.695 >> 2star O2 176 12.421 23.171 16.695 >> 2star O2 177 12.421 23.601 16.695 >> 2star O2 178 12.421 24.031 16.695 >> 2star O2 179 12.421 24.461 16.695 >> 2star O2 180 12.421 24.891 16.695 >> 2star O2 181 12.421 25.321 16.695 >> 2star O2 182 12.421 25.751 16.695 >> 2star O2 183 12.421 26.181 16.695 >> >> Notice the jump on atom number 174 and 175, why is there a sudden coordinate >> jump? >> All i can see in VMD is my negative axis part of the molecule beads are cut >> off. >> > > editconf builds boxes from the coordinate origin so that there are never > negative coordinates. It should be able to deal with those coordinates, > equally shifted, which it appears they were. Can you provide images of > what's going on? I don't clearly understand what's wrong. What do you mean > by "cut off?" Nonexistent? Incorrectly placed? > > -Justin > > -- > ======================================== > > Justin A. Lemkul, Ph.D. > Research Scientist > Department of Biochemistry > Virginia Tech > Blacksburg, VA > jalemkul[at]vt.edu | (540) 231-9080 > http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin > > ======================================== > -- > gmx-users mailing list gmx-users@gromacs.org > http://lists.gromacs.org/mailman/listinfo/gmx-users > * Please search the archive at > http://www.gromacs.org/Support/Mailing_Lists/Search before posting! > * Please don't post (un)subscribe requests to the list. Use the www interface > or send it to gmx-users-requ...@gromacs.org. > * Can't post? Read http://www.gromacs.org/Support/Mailing_Lists -- gmx-users mailing list gmx-users@gromacs.org http://lists.gromacs.org/mailman/listinfo/gmx-users * Please search the archive at http://www.gromacs.org/Support/Mailing_Lists/Search before posting! * Please don't post (un)subscribe requests to the list. Use the www interface or send it to gmx-users-requ...@gromacs.org. * Can't post? Read http://www.gromacs.org/Support/Mailing_Lists