Hi Justin! Do you maybe have an example of such a protein (preferably not too large :-), that I could run some tests on?
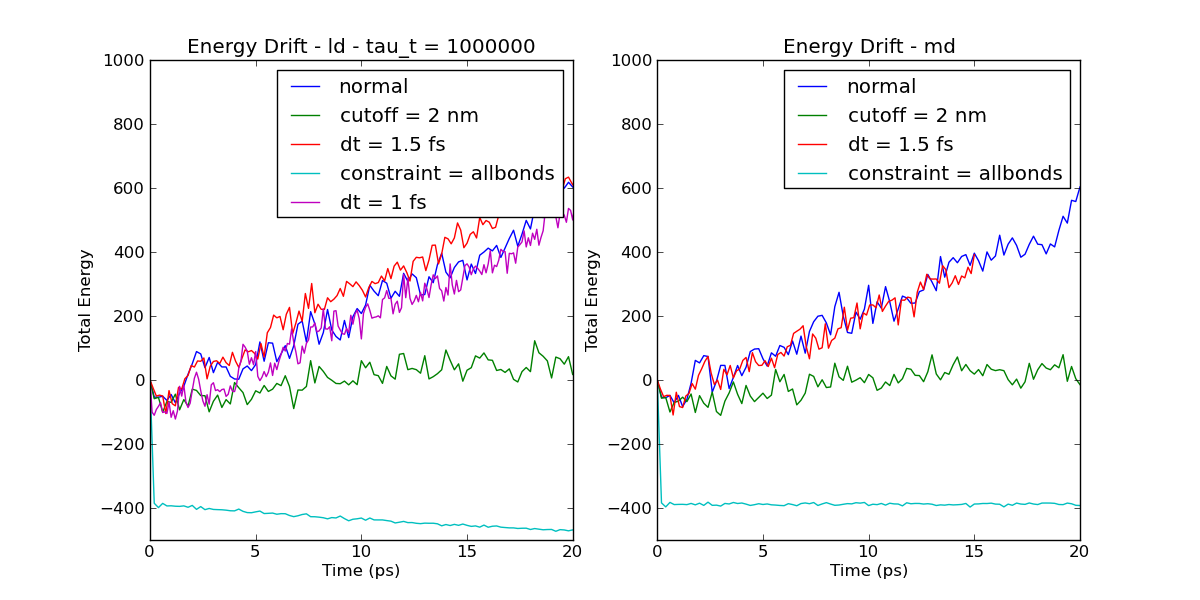
I'd be interested in seeing if there has been any bugs introduced in the cutoff code that destabilises proteins that way. Thanks /Per 18 okt 2011 kl. 23:41 skrev "Justin A. Lemkul" <jalem...@vt.edu>: > > > Ben Reynwar wrote: >> I posted to the list a few days ago with an energy drift problem. >> Mark Abraham helpfully suggested using all-bonds rather than h-bonds >> which solved the problem. I'm now trying to understand quite why that >> helped so much. >> The simulation is a protein of about 5000 atoms using GBSA, a time >> step of 2 fs, and a cut-off of 1.6 for VdW, coulomb and GB. >> I've run energy drift simulations using the md integrator, with no >> thermostat, and using the ld integrator with a tau_t of 1000000. >> Simulations were run changing various parameters. The changes were: >> - dt reduced to 1.5 >> - dt reduced to 1.0 >> - cutoff increased to 2.0 >> - constraint changed from h-bonds to all-bonds >> Plot of the energy drifts can be seen at >> http://www.reynwar.net/ben/gromacs/energy_drift.png. >> Noticeable features are: >> - changing the time step makes no difference (in my last post I >> claimed it did, which is why you should make plots rather than >> eyeballing log files). >> - increasing the cut-off helps a lot. >> - changing constraint to all-bonds make a dramatic difference >> - using ld there is a downwards drift in the energy when using >> all-bonds constraint. The temperature is roughly 300 K and the set >> point is 400 K so this downwards drift seems unlikely to be due to >> coupling to the langevin thermostat. >> My questions are: >> - why does the all-bonds constraint help so much? >> - why doesn't moving to a smaller time step help with this? >> - what is the cause of the downwards drift when using ld with all-bonds? > > I can offer a bit of general advice here, but no specific answers. Hopefully > it helps. To get at the root cause of all of this, you should be analyzing > individual energy components, not just the total energy. These will tell you > which terms are systematically changing. > > In my experience with GB simulations, using anything other than infinite > cutoffs (i.e. all-vs-all kernel) has resulted in unstable simulations. I > don't mean that the simulations crash or anything, but the results are > clearly incorrect. Stably folded proteins have drastically increased RMSD and > lose their structure very easily. The effect is independent of the chosen > force field. I would recommend always using infinite cutoffs for GB > simulations. It may improve your situation. > > -Justin > > -- > ======================================== > > Justin A. Lemkul > Ph.D. Candidate > ICTAS Doctoral Scholar > MILES-IGERT Trainee > Department of Biochemistry > Virginia Tech > Blacksburg, VA > jalemkul[at]vt.edu | (540) 231-9080 > http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin > > ======================================== > -- > gmx-users mailing list gmx-users@gromacs.org > http://lists.gromacs.org/mailman/listinfo/gmx-users > Please search the archive at > http://www.gromacs.org/Support/Mailing_Lists/Search before posting! > Please don't post (un)subscribe requests to the list. Use the www interface > or send it to gmx-users-requ...@gromacs.org. > Can't post? Read http://www.gromacs.org/Support/Mailing_Lists -- gmx-users mailing list gmx-users@gromacs.org http://lists.gromacs.org/mailman/listinfo/gmx-users Please search the archive at http://www.gromacs.org/Support/Mailing_Lists/Search before posting! Please don't post (un)subscribe requests to the list. Use the www interface or send it to gmx-users-requ...@gromacs.org. Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
{kind=link}