Ben Reynwar wrote:
I posted to the list a few days ago with an energy drift problem.
Mark Abraham helpfully suggested using all-bonds rather than h-bonds
which solved the problem. I'm now trying to understand quite why that
helped so much.
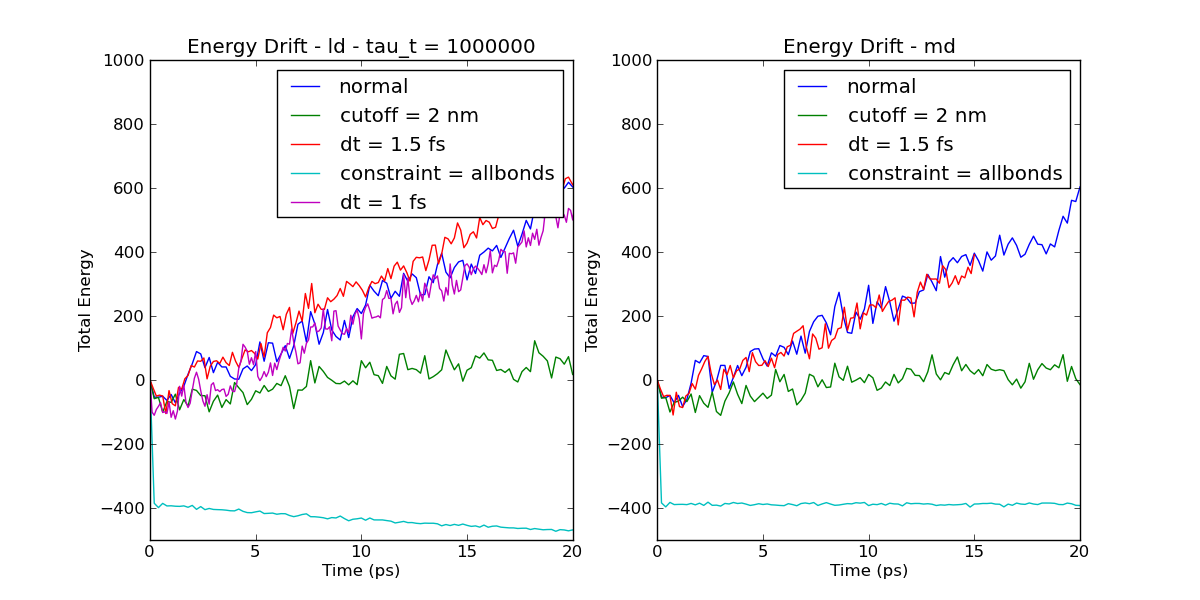
The simulation is a protein of about 5000 atoms using GBSA, a time
step of 2 fs, and a cut-off of 1.6 for VdW, coulomb and GB.
I've run energy drift simulations using the md integrator, with no
thermostat, and using the ld integrator with a tau_t of 1000000.
Simulations were run changing various parameters. The changes were:
- dt reduced to 1.5
- dt reduced to 1.0
- cutoff increased to 2.0
- constraint changed from h-bonds to all-bonds
Plot of the energy drifts can be seen at
http://www.reynwar.net/ben/gromacs/energy_drift.png.
Noticeable features are:
- changing the time step makes no difference (in my last post I
claimed it did, which is why you should make plots rather than
eyeballing log files).
- increasing the cut-off helps a lot.
- changing constraint to all-bonds make a dramatic difference
- using ld there is a downwards drift in the energy when using
all-bonds constraint. The temperature is roughly 300 K and the set
point is 400 K so this downwards drift seems unlikely to be due to
coupling to the langevin thermostat.
My questions are:
- why does the all-bonds constraint help so much?
- why doesn't moving to a smaller time step help with this?
- what is the cause of the downwards drift when using ld with all-bonds?
I can offer a bit of general advice here, but no specific answers. Hopefully it
helps. To get at the root cause of all of this, you should be analyzing
individual energy components, not just the total energy. These will tell you
which terms are systematically changing.
In my experience with GB simulations, using anything other than infinite cutoffs
(i.e. all-vs-all kernel) has resulted in unstable simulations. I don't mean
that the simulations crash or anything, but the results are clearly incorrect.
Stably folded proteins have drastically increased RMSD and lose their structure
very easily. The effect is independent of the chosen force field. I would
recommend always using infinite cutoffs for GB simulations. It may improve your
situation.
-Justin
--
========================================
Justin A. Lemkul
Ph.D. Candidate
ICTAS Doctoral Scholar
MILES-IGERT Trainee
Department of Biochemistry
Virginia Tech
Blacksburg, VA
jalemkul[at]vt.edu | (540) 231-9080
http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin
========================================
--
gmx-users mailing list gmx-users@gromacs.org
http://lists.gromacs.org/mailman/listinfo/gmx-users
Please search the archive at
http://www.gromacs.org/Support/Mailing_Lists/Search before posting!
Please don't post (un)subscribe requests to the list. Use the
www interface or send it to gmx-users-requ...@gromacs.org.
Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
{kind=link}