Randy Read's paper in latest Acta D: Measuring and using information gained by observing diffraction data http://journals.iucr.org/d/issues/2020/03/00/ba5308/index.html
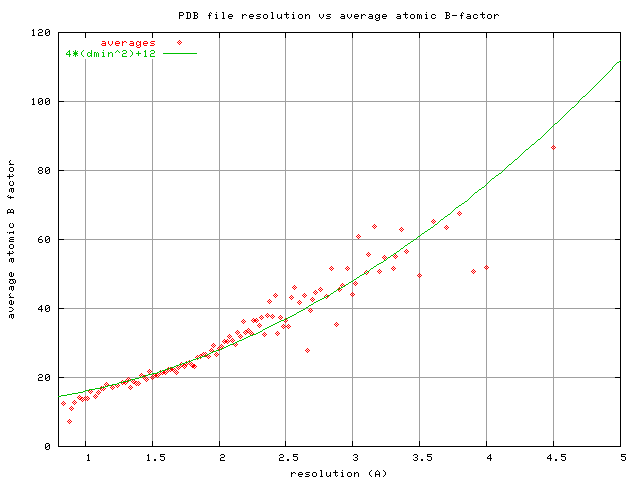
seems very relevant to this discussion! Pavel On Fri, Mar 6, 2020 at 8:44 AM James Holton <[email protected]> wrote: > Thank you Kay, > > Very good points, as always. I was thinking there must be a better > apodization filter than cutoffs and B factors. I'll have to try a > CC1/2-based roll-off. But, I wonder if this could be done better on a > per-reflection basis? Taking advantage of the sigmas? I have tried > using 1/sigma^2 as a weight in map calculation, and that makes the map > look really weird. Your idea makes more sense. > > On the other hand, the French-Wilson (F&W) truncation procedure is > supposed to come up with the maximum-likelihood Fourier coefficient > given the observed intensity and sigma(intensity). So, the "F" values > we get from truncate or xdsconv should already be "good"? Maybe the > problem is that we are sharpening after the F&W step, rather than > before? Or maybe the problem is that F&W bottoms out around F=sig(F). > Your proposed weight might finish the job... > > As for sigmaA, one thing I do know is that refmac5 now uses experimental > sigmas by default. Phenix.refine does not. > > One thing is for sure, sharpening the data before refinement is not > going to become a popular strategy. This is because pre-sharpened data > will make Rwork and Rfree higher than they are with the "natural" B > factor. You can also make your Rwork Rfree much lower by applying a > positive B factor to your data before starting refinement. This comes > with absolutely no improvement in model quality, so please don't try > this at home. > > -James Holton > MAD Scientist > > On 3/5/2020 9:38 PM, Kay Diederichs wrote: > > Dear James, > > > > important and educational points! This triggers some thoughts ... > > > > The one point where I don't quite agree is with "What about filtering > out the noise? An ideal noise suppression filter has the same shape as the > signal (I found that in Numerical Recipes), and the shape of the signal > from a macromolecule is a Gaussian in reciprocal space (aka straight line > on a Wilson plot). This is true, by the way, for both a molecule packed > into a crystal or free in solution. So, the ideal noise-suppression filter > is simply applying a B factor. " > > > > I think we can do better than that. We should use the knowledge about > the actual signal and its noise (which we measure) as a weighting factor, > rather than that of the theoretical signal (the straight line in the Wilson > plot), for the purpose of noise suppression. Formula 13.3.6 in Numerical > Recipes (3rd ed., 2007), which gives the optimal (Wiener) filter to be used > for weighting, is phi(f) = S(f)^2/(S(f)^2 + N(f)^2) . But at high > resolution, this is just CC1/2: see Box 1 of reference (1) - the formula > CC1/2 = 1/(1 + 2/(I/sigma)^2 can be written as CC1/2 = <I>^2/(<I>^2 + 2 > <sigma>^2) where sigma is the estimate of the noise in I ; don't know right > now why there is a factor of 2). This goes to zero at the resolution where > the signal goes to zero, and is near one in the resolution range in which > we have good knowledge of the signal. > > (I only thought about this today, and I also considered CC* as a > weighting factor, as I understand is suggested by Rosenthal and Henderson, > J.Mol.Biol. 2003, but I cannot convince myself currently that this is > right. Anyway, the shape of the CC* curve as a function of resolution > matches that of CC1/2) > > In other words, we should be able to suppress the noise by multiplying > the Fourier coefficients used for map calculation with (a smooth > resolution-dependent approximation of) CC1/2. This should allow to sharpen, > with the best noise suppression we can get. > > > > Thinking about this, we are already typically using weighted Fourier > coefficients of the form 2mFobs-DFcalc for map calculation. Aren't these > already weighted in the correct way? I think not - those m and D weights > are calculated from estimates of model (in-)accuracy and (in-)completeness, > but don't properly take the measurement errors into account. Of course, > since noisy data make the sigmaA values worse, the noise in the data > influences sigmaA, but not in the functionally correct form. To my > understanding, the correct way to take account of both model and data > errors is given by reference (2), which - to my knowledge - is not yet > implemented except in PHASER. > > > > Hope this makes sense! > > > > Kay > > > > References: > > (1) Karplus & Diederichs (2015) Assessing and maximizing data quality in > macromolecular crystallography. > > Curr. Opin. Struct. Biol. 34, 60-68 . PDF at > https://www.biologie.uni-konstanz.de/typo3temp/secure_downloads/82815/0/2b10c9e6f9a28129e1b119d21aeeab217c918bb1/Karplus2015_CurrOpinStructBiol.pdf > > (2) RJ Read, AJ McCoy (2016) A log-likelihood-gain intensity target for > crystallographic phasing that accounts for experimental error. Acta > Crystallographica Section D: Structural Biology 72 (3), 375-387 > > https://scripts.iucr.org/cgi-bin/paper?dz5382 > > > > On Thu, 5 Mar 2020 01:11:33 +0100, James Holton <[email protected]> > wrote: > > > >> The funny thing is, although we generally regard resolution as a primary > >> indicator of data quality the appearance of a density map at the classic > >> "1-sigma" contour has very little to do with resolution, and everything > >> to do with the B factor. > >> > >> Seriously, try it. Take any structure you like, set all the B factors to > >> 30 with PDBSET, calculate a map with SFALL or phenix.fmodel and have a > >> look at the density of tyrosine (Tyr) side chains. Even if you > >> calculate structure factors all the way out to 1.0 A the holes in the > >> Tyr rings look exactly the same: just barely starting to form. This is > >> because the structure factors from atoms with B=30 are essentially zero > >> out at 1.0 A, and adding zeroes does not change the map. You can adjust > >> the contour level, of course, and solvent content will have some effect > >> on where the "1-sigma" contour lies, but generally B=30 is the point > >> where Tyr side chains start to form their holes. Traditionally, this is > >> attributed to 1.8A resolution, but it is really at B=30. The point > >> where waters first start to poke out above the 1-sigma contour is at > >> B=60, despite being generally attributed to d=2.7A. > >> > >> Now, of course, if you cut off this B=30 data at 3.5A then the Tyr side > >> chains become blobs, but that is equivalent to collecting data with the > >> detector way too far away and losing your high-resolution spots off the > >> edges. I have seen a few people do that, but not usually for a > >> published structure. Most people fight very hard for those faint, > >> barely-existing high-angle spots. But why do we do that if the map is > >> going to look the same anyway? The reason is because resolution and B > >> factors are linked. > >> > >> Resolution is about separation vs width, and the width of the density > >> peak from any atom is set by its B factor. Yes, atoms have an intrinsic > >> width, but it is very quickly washed out by even modest B factors (B > > >> 10). This is true for both x-ray and electron form factors. To a very > >> good approximation, the FWHM of C, N and O atoms is given by: > >> FWHM= sqrt(B*log(2))/pi+0.15 > >> > >> where "B" is the B factor assigned to the atom and the 0.15 fudge factor > >> accounts for its intrinsic width when B=0. Now that we know the peak > >> width, we can start to ask if two peaks are "resolved". > >> > >> Start with the classical definition of "resolution" (call it after Airy, > >> Raleigh, Dawes, or whatever famous person you like), but essentially you > >> are asking the question: "how close can two peaks be before they merge > >> into one peak?". For Gaussian peaks this is 0.849*FWHM. Simple enough. > >> However, when you look at the density of two atoms this far apart you > >> will see the peak is highly oblong. Yes, the density has one maximum, > >> but there are clearly two atoms in there. It is also pretty obvious the > >> long axis of the peak is the line between the two atoms, and if you fit > >> two round atoms into this peak you recover the distance between them > >> quite accurately. Are they really not "resolved" if it is so clear > >> where they are? > >> > >> In such cases you usually want to sharpen, as that will make the oblong > >> blob turn into two resolved peaks. Sharpening reduces the B factor and > >> therefore FWHM of every atom, making the "resolution" (0.849*FWHM) a > >> shorter distance. So, we have improved resolution with sharpening! Why > >> don't we always do this? Well, the reason is because of noise. > >> Sharpening up-weights the noise of high-order Fourier terms and > >> therefore degrades the overall signal-to-noise (SNR) of the map. This > >> is what I believe Colin would call reduced "contrast". Of course, since > >> we view maps with a threshold (aka contour) a map with SNR=5 will look > >> almost identical to a map with SNR=500. The "noise floor" is generally > >> well below the 1-sigma threshold, or even the 0-sigma threshold > >> (https://doi.org/10.1073/pnas.1302823110). As you turn up the > >> sharpening you will see blobs split apart and also see new peaks rising > >> above your map contouring threshold. Are these new peaks real? Or are > >> they noise? That is the difference between SNR=500 and SNR=5, > >> respectively. The tricky part of sharpening is knowing when you have > >> reached the point where you are introducing more noise than signal. > >> There are some good methods out there, but none of them are perfect. > >> > >> What about filtering out the noise? An ideal noise suppression filter > >> has the same shape as the signal (I found that in Numerical Recipes), > >> and the shape of the signal from a macromolecule is a Gaussian in > >> reciprocal space (aka straight line on a Wilson plot). This is true, by > >> the way, for both a molecule packed into a crystal or free in solution. > >> So, the ideal noise-suppression filter is simply applying a B factor. > >> Only problem is: sharpening is generally done by applying a negative B > >> factor, so applying a Gaussian blur is equivalent to just not sharpening > >> as much. So, we are back to "optimal sharpening" again. > >> > >> Why not use a filter that is non-Gaussian? We do this all the time! > >> Cutting off the data at a given resolution (d) is equivalent to blurring > >> the map with this function: > >> > >> kernel_d(r) = 4/3*pi/d**3*sinc3(2*pi*r/d) > >> sinc3(x) = (x==0?1:3*(sin(x)/x-cos(x))/(x*x)) > >> > >> where kernel_d(r) is the normalized weight given to a point "r" Angstrom > >> away from the center of each blurring operation, and "sinc3" is the > >> Fourier synthesis of a solid sphere. That is, if you make an HKL file > >> with all F=1 and PHI=0 out to a resolution d, then effectively all hkls > >> beyond the resolution limit are zero. If you calculate a map with those > >> Fs, you will find the kernel_d(r) function at the origin. What that > >> means is: by applying a resolution cutoff, you are effectively > >> multiplying your data by this sphere of unit Fs, and since a > >> multiplication in reciprocal space is a convolution in real space, the > >> effect is convoluting (blurring) with kernel_d(x). > >> > >> For comparison, if you apply a B factor, the real-space blurring kernel > >> is this: > >> kernel_B(r) = (4*pi/B)**1.5*exp(-4*pi**2/B*r*r) > >> > >> If you graph these two kernels (format is for gnuplot) you will find > >> that they have the same FWHM whenever B=80*(d/3)**2. This "rule" is the > >> one I used for my resolution demonstration movie I made back in the late > >> 20th century: > >> https://bl831.als.lbl.gov/~jamesh/movies/index.html#resolution > >> > >> What I did then was set all atomic B factors to B = 80*(d/3)^2 and then > >> cut the resolution at "d". Seemed sensible at the time. I suppose I > >> could have used the PDB-wide average atomic B factor reported for > >> structures with resolution "d", which roughly follows: > >> B = 4*d**2+12 > >> https://bl831.als.lbl.gov/~jamesh/pickup/reso_vs_avgB.png > >> > >> The reason I didn't use this formula for the movie is because I didn't > >> figure it out until about 10 years later. These two curves cross at > >> 1.5A, but diverge significantly at poor resolution. So, which one is > >> right? It depends on how well you can measure really really faint > >> spots, and we've been getting better at that in recent decades. > >> > >> So, what I'm trying to say here is that just because your data has CC1/2 > >> or FSC dropping off to insignificance at 1.8 A doesn't mean you are > >> going to see holes in Tyr side chains. However, if you measure your > >> weak, high-res data really well (high multiplicity), you might be able > >> to sharpen your way to a much clearer map. > >> > >> -James Holton > >> MAD Scientist > >> > >> On 2/27/2020 11:01 AM, Nave, Colin (DLSLtd,RAL,LSCI) wrote: > >>> James > >>> > >>> All you say seems sensible to me but there is the possibility of > >>> confusion regarding the use of the word threshold. I fully agree that > >>> a half bit information threshold is inappropriate if it is taken to > >>> mean that the data should be truncated at that resolution. The ever > >>> more sophisticated refinement programs are becoming adept at handling > >>> the noisy data. > >>> > >>> The half bit information threshold I was discussing refers to a > >>> nominal resolution. This is not just for trivial reporting purposes. > >>> The half bit threshold is being used to compare imaging methods and > >>> perhaps demonstrate that significant information is present with a > >>> dose below any radiation damage threshold (that word again). The > >>> justification for doing this appears to come from the fact it has been > >>> adopted for protein structure determination by single particle > >>> electron microscopy. However, low contrast features might not be > >>> visible at this nominal resolution. > >>> > >>> The analogy with protein crystallography might be to collect data > >>> below an absorption edge to give a nominal resolution of 2 angstrom. > >>> Then do it again well above the absorption edge. The second one gives > >>> much greater Bijvoet differences despite the fact that the nominal > >>> resolution is the same. I doubt whether anyone doing this would be > >>> misled by this as they would examine the statistics for the Bijvoet > >>> differences instead. However, it does indicate the relationship > >>> between contrast and resolution. > >>> > >>> The question, if referring to an information threshold for nominal > >>> resolution, could be “Is there significant information in the data at > >>> the required contrast and resolution?”. Then “Can one obtain this > >>> information at a dose below any radiation damage limit” > >>> > >>> Keep posting! > >>> > >>> Regards > >>> > >>> Colin > >>> > >>> *From:*James Holton <[email protected]> > >>> *Sent:* 27 February 2020 01:14 > >>> *To:* [email protected] > >>> *Cc:* Nave, Colin (DLSLtd,RAL,LSCI) <[email protected]> > >>> *Subject:* Re: [ccp4bb] [3dem] Which resolution? > >>> > >>> In my opinion the threshold should be zero bits. Yes, this is where > >>> CC1/2 = 0 (or FSC = 0). If there is correlation then there is > >>> information, and why throw out information if there is information to > >>> be had? Yes, this information comes with noise attached, but that is > >>> why we have weights. > >>> > >>> It is also important to remember that zero intensity is still useful > >>> information. Systematic absences are an excellent example. They have > >>> no intensity at all, but they speak volumes about the structure. In a > >>> similar way, high-angle zero-intensity observations also tell us > >>> something. Ever tried unrestrained B factor refinement at poor > >>> resolution? It is hard to do nowadays because of all the safety > >>> catches in modern software, but you can get great R factors this way. > >>> A telltale sign of this kind of "over fitting" is remarkably large > >>> Fcalc values beyond the resolution cutoff. These don't contribute to > >>> the R factor, however, because Fobs is missing for these hkls. So, > >>> including zero-intensity data suppresses at least some types of > >>> over-fitting. > >>> > >>> The thing I like most about the zero-information resolution cutoff is > >>> that it forces us to address the real problem: what do you mean by > >>> "resolution" ? Not long ago, claiming your resolution was 3.0 A meant > >>> that after discarding all spots with individual I/sigI < 3 you still > >>> have 80% completeness in the 3.0 A bin. Now we are saying we have a > >>> 3.0 A data set when we can prove statistically that a few > >>> non-background counts fell into the sum of all spot areas at 3.0 A. > >>> These are not the same thing. > >>> > >>> Don't get me wrong, including the weak high-resolution information > >>> makes the model better, and indeed I am even advocating including all > >>> the noisy zeroes. However, weak data at 3.0 A is never going to be as > >>> good as having strong data at 3.0 A. So, how do we decide? I > >>> personally think that the resolution assigned to the PDB deposition > >>> should remain the classical I/sigI > 3 at 80% rule. This is really > >>> the only way to have meaningful comparison of resolution between very > >>> old and very new structures. One should, of course, deposit all the > >>> data, but don't claim that cut-off as your "resolution". That is just > >>> plain unfair to those who came before. > >>> > >>> Oh yeah, and I also have a session on "interpreting low-resolution > >>> maps" at the GRC this year. > >>> > https://www.grc.org/diffraction-methods-in-structural-biology-conference/2020/ > >>> > >>> So, please, let the discussion continue! > >>> > >>> -James Holton > >>> MAD Scientist > >>> > >>> On 2/22/2020 11:06 AM, Nave, Colin (DLSLtd,RAL,LSCI) wrote: > >>> > >>> Alexis > >>> > >>> This is a very useful summary. > >>> > >>> You say you were not convinced by Marin's derivation in 2005. Are > >>> you convinced now and, if not, why? > >>> > >>> My interest in this is that the FSC with half bit thresholds have > >>> the danger of being adopted elsewhere because they are becoming > >>> standard for protein structure determination (by EM or MX). If it > >>> is used for these mature techniques it must be right! > >>> > >>> It is the adoption of the ½ bit threshold I worry about. I gave a > >>> rather weak example for MX which consisted of partial occupancy of > >>> side chains, substrates etc. For x-ray imaging a wide range of > >>> contrasts can occur and, if you want to see features with only a > >>> small contrast above the surroundings then I think the half bit > >>> threshold would be inappropriate. > >>> > >>> It would be good to see a clear message from the MX and EM > >>> communities as to why an information content threshold of ½ a bit > >>> is generally appropriate for these techniques and an > >>> acknowledgement that this threshold is technique/problem > dependent. > >>> > >>> We might then progress from the bronze age to the iron age. > >>> > >>> Regards > >>> > >>> Colin > >>> > >>> *From:*CCP4 bulletin board <[email protected]> > >>> <mailto:[email protected]> *On Behalf Of *Alexis Rohou > >>> *Sent:* 21 February 2020 16:35 > >>> *To:* [email protected] <mailto:[email protected]> > >>> *Subject:* Re: [ccp4bb] [3dem] Which resolution? > >>> > >>> Hi all, > >>> > >>> For those bewildered by Marin's insistence that everyone's been > >>> messing up their stats since the bronze age, I'd like to offer > >>> what my understanding of the situation. More details in this > >>> thread from a few years ago on the exact same topic: > >>> > >>> > https://mail.ncmir.ucsd.edu/pipermail/3dem/2015-August/003939.html > >>> > >>> > https://mail.ncmir.ucsd.edu/pipermail/3dem/2015-August/003944.html > >>> > >>> Notwithstanding notational problems (e.g. strict equations as > >>> opposed to approximation symbols, or omission of symbols to denote > >>> estimation), I believe Frank & Al-Ali and "descendent" papers > >>> (e.g. appendix of Rosenthal & Henderson 2003) are fine. The cross > >>> terms that Marin is agitated about indeed do in fact have an > >>> expectation value of 0.0 (in the ensemble; if the experiment were > >>> performed an infinite number of times with different realizations > >>> of noise). I don't believe Pawel or Jose Maria or any of the other > >>> authors really believe that the cross-terms are orthogonal. > >>> > >>> When N (the number of independent Fouier voxels in a shell) is > >>> large enough, mean(Signal x Noise) ~ 0.0 is only an approximation, > >>> but a pretty good one, even for a single FSC experiment. This is > >>> why, in my book, derivations that depend on Frank & Al-Ali are OK, > >>> under the strict assumption that N is large. Numerically, this > >>> becomes apparent when Marin's half-bit criterion is plotted - > >>> asymptotically it has the same behavior as a constant threshold. > >>> > >>> So, is Marin wrong to worry about this? No, I don't think so. > >>> There are indeed cases where the assumption of large N is broken. > >>> And under those circumstances, any fixed threshold (0.143, 0.5, > >>> whatever) is dangerous. This is illustrated in figures of van Heel > >>> & Schatz (2005). Small boxes, high-symmetry, small objects in > >>> large boxes, and a number of other conditions can make fixed > >>> thresholds dangerous. > >>> > >>> It would indeed be better to use a non-fixed threshold. So why am > >>> I not using the 1/2-bit criterion in my own work? While > >>> numerically it behaves well at most resolution ranges, I was not > >>> convinced by Marin's derivation in 2005. Philosophically though, I > >>> think he's right - we should aim for FSC thresholds that are more > >>> robust to the kinds of edge cases mentioned above. It would be the > >>> right thing to do. > >>> > >>> Hope this helps, > >>> > >>> Alexis > >>> > >>> On Sun, Feb 16, 2020 at 9:00 AM Penczek, Pawel A > >>> <[email protected] <mailto:[email protected] > >> > >>> wrote: > >>> > >>> Marin, > >>> > >>> The statistics in 2010 review is fine. You may disagree with > >>> assumptions, but I can assure you the “statistics” (as you > >>> call it) is fine. Careful reading of the paper would reveal to > >>> you this much. > >>> > >>> Regards, > >>> > >>> Pawel > >>> > >>> > >>> > >>> > >>> On Feb 16, 2020, at 10:38 AM, Marin van Heel > >>> <[email protected] > >>> <mailto:[email protected]>> wrote: > >>> > >>> > >>> > >>> ***** EXTERNAL EMAIL ***** > >>> > >>> Dear Pawel and All others .... > >>> > >>> This 2010 review is - unfortunately - largely based on the > >>> flawed statistics I mentioned before, namely on the a > >>> priori assumption that the inner product of a signal > >>> vector and a noise vector are ZERO (an orthogonality > >>> assumption). The (Frank & Al-Ali 1975) paper we have > >>> refuted on a number of occasions (for example in 2005, and > >>> most recently in our BioRxiv paper) but you still take > >>> that as the correct relation between SNR and FRC (and you > >>> never cite the criticism...). > >>> > >>> Sorry > >>> > >>> Marin > >>> > >>> On Thu, Feb 13, 2020 at 10:42 AM Penczek, Pawel A > >>> <[email protected] > >>> <mailto:[email protected]>> wrote: > >>> > >>> Dear Teige, > >>> > >>> I am wondering whether you are familiar with > >>> > >>> > >>> Resolution measures in molecular electron > microscopy. > >>> > >>> Penczek PA. Methods Enzymol. 2010. > >>> > >>> > >>> Citation > >>> > >>> Methods Enzymol. 2010;482:73-100. doi: > >>> 10.1016/S0076-6879(10)82003-8. > >>> > >>> You will find there answers to all questions you asked > >>> and much more. > >>> > >>> Regards, > >>> > >>> Pawel Penczek > >>> > >>> Regards, > >>> > >>> Pawel > >>> > >>> _______________________________________________ > >>> 3dem mailing list > >>> [email protected] <mailto:[email protected]> > >>> https://mail.ncmir.ucsd.edu/mailman/listinfo/3dem > >>> < > https://urldefense.proofpoint.com/v2/url?u=https-3A__mail.ncmir.ucsd.edu_mailman_listinfo_3dem&d=DwMFaQ&c=bKRySV-ouEg_AT-w2QWsTdd9X__KYh9Eq2fdmQDVZgw&r=yEYHb4SF2vvMq3W-iluu41LlHcFadz4Ekzr3_bT4-qI&m=3-TZcohYbZGHCQ7azF9_fgEJmssbBksaI7ESb0VIk1Y&s=XHMq9Q6Zwa69NL8kzFbmaLmZA9M33U01tBE6iAtQ140&e= > > > >>> > >>> _______________________________________________ > >>> 3dem mailing list > >>> [email protected] <mailto:[email protected]> > >>> https://mail.ncmir.ucsd.edu/mailman/listinfo/3dem > >>> > >>> > ------------------------------------------------------------------------ > >>> > >>> To unsubscribe from the CCP4BB list, click the following link: > >>> https://www.jiscmail.ac.uk/cgi-bin/webadmin?SUBED1=CCP4BB&A=1 > >>> > >>> -- > >>> > >>> This e-mail and any attachments may contain confidential, > >>> copyright and or privileged material, and are for the use of the > >>> intended addressee only. If you are not the intended addressee or > >>> an authorised recipient of the addressee please notify us of > >>> receipt by returning the e-mail and do not use, copy, retain, > >>> distribute or disclose the information in or attached to the > e-mail. > >>> Any opinions expressed within this e-mail are those of the > >>> individual and not necessarily of Diamond Light Source Ltd. > >>> Diamond Light Source Ltd. cannot guarantee that this e-mail or any > >>> attachments are free from viruses and we cannot accept liability > >>> for any damage which you may sustain as a result of software > >>> viruses which may be transmitted in or with the message. > >>> Diamond Light Source Limited (company no. 4375679). Registered in > >>> England and Wales with its registered office at Diamond House, > >>> Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 > >>> 0DE, United Kingdom > >>> > >>> > ------------------------------------------------------------------------ > >>> > >>> To unsubscribe from the CCP4BB list, click the following link: > >>> https://www.jiscmail.ac.uk/cgi-bin/webadmin?SUBED1=CCP4BB&A=1 > >>> > >>> -- > >>> > >>> This e-mail and any attachments may contain confidential, copyright > >>> and or privileged material, and are for the use of the intended > >>> addressee only. If you are not the intended addressee or an authorised > >>> recipient of the addressee please notify us of receipt by returning > >>> the e-mail and do not use, copy, retain, distribute or disclose the > >>> information in or attached to the e-mail. > >>> Any opinions expressed within this e-mail are those of the individual > >>> and not necessarily of Diamond Light Source Ltd. > >>> Diamond Light Source Ltd. cannot guarantee that this e-mail or any > >>> attachments are free from viruses and we cannot accept liability for > >>> any damage which you may sustain as a result of software viruses which > >>> may be transmitted in or with the message. > >>> Diamond Light Source Limited (company no. 4375679). Registered in > >>> England and Wales with its registered office at Diamond House, Harwell > >>> Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, United > >>> Kingdom > >>> > >> > >> ######################################################################## > >> > >> To unsubscribe from the CCP4BB list, click the following link: > >> https://www.jiscmail.ac.uk/cgi-bin/webadmin?SUBED1=CCP4BB&A=1 > >> > > ######################################################################## > > > > To unsubscribe from the CCP4BB list, click the following link: > > https://www.jiscmail.ac.uk/cgi-bin/webadmin?SUBED1=CCP4BB&A=1 > > ######################################################################## > > To unsubscribe from the CCP4BB list, click the following link: > https://www.jiscmail.ac.uk/cgi-bin/webadmin?SUBED1=CCP4BB&A=1 > ######################################################################## To unsubscribe from the CCP4BB list, click the following link: https://www.jiscmail.ac.uk/cgi-bin/webadmin?SUBED1=CCP4BB&A=1
{kind=link}