James Starlight wrote:
Justin,
2011/11/11 Justin A. Lemkul <jalem...@vt.edu <mailto:jalem...@vt.edu>>
If the bilayer is splitting across periodic boundaries (I'm assuming
that's what you mean by "pb"), then it's more likely that you placed
the components of your system in an inconvenient way than it is that
something is actually wrong.
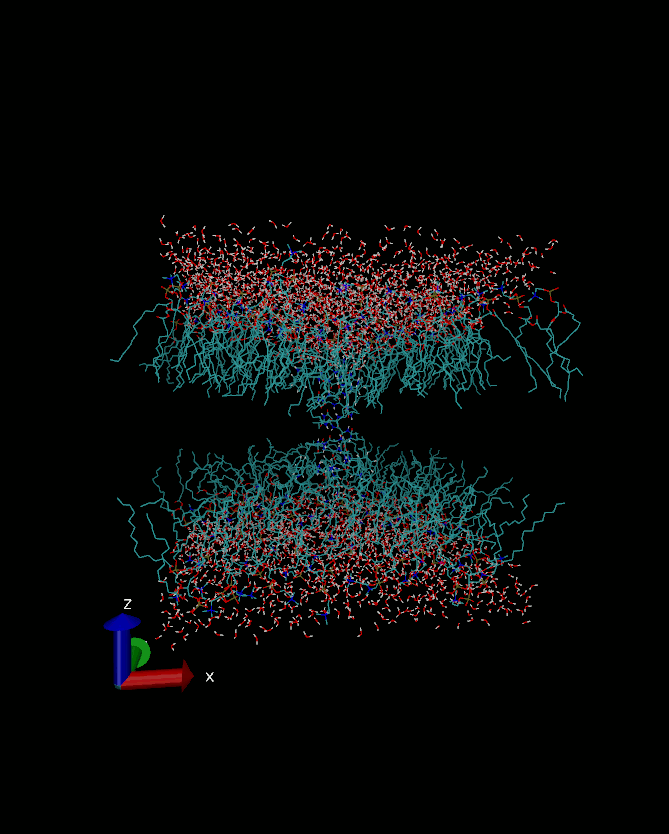
Bilayer has moved on Z-dimension in periodic box. That is actually
what's happend :)
http://i1209.photobucket.com/albums/cc394/own11/vmdscene.gif
How large of a void exists between your lipids and water prior to starting the
simulation? I'm almost certain that you built the unit cell incorrectly - there
should never be that much free space.
By the way I'ts intresting that Gmembed has removed 6 lipids in
comparison to the inflateglo ( althought I've done all things in
accordance to the KALP tutorial because my protein is the same).
Different algorithms, different results. Nothing odd about that to me.
-Justin
--
========================================
Justin A. Lemkul
Ph.D. Candidate
ICTAS Doctoral Scholar
MILES-IGERT Trainee
Department of Biochemistry
Virginia Tech
Blacksburg, VA
jalemkul[at]vt.edu | (540) 231-9080
http://www.bevanlab.biochem.vt.edu/Pages/Personal/justin
========================================
--
gmx-users mailing list gmx-users@gromacs.org
http://lists.gromacs.org/mailman/listinfo/gmx-users
Please search the archive at
http://www.gromacs.org/Support/Mailing_Lists/Search before posting!
Please don't post (un)subscribe requests to the list. Use the
www interface or send it to gmx-users-requ...@gromacs.org.
Can't post? Read http://www.gromacs.org/Support/Mailing_Lists
{kind=link}