Dear CCP4ers, I'd like to kindly ask your advice. Sorry for the long e-mail.
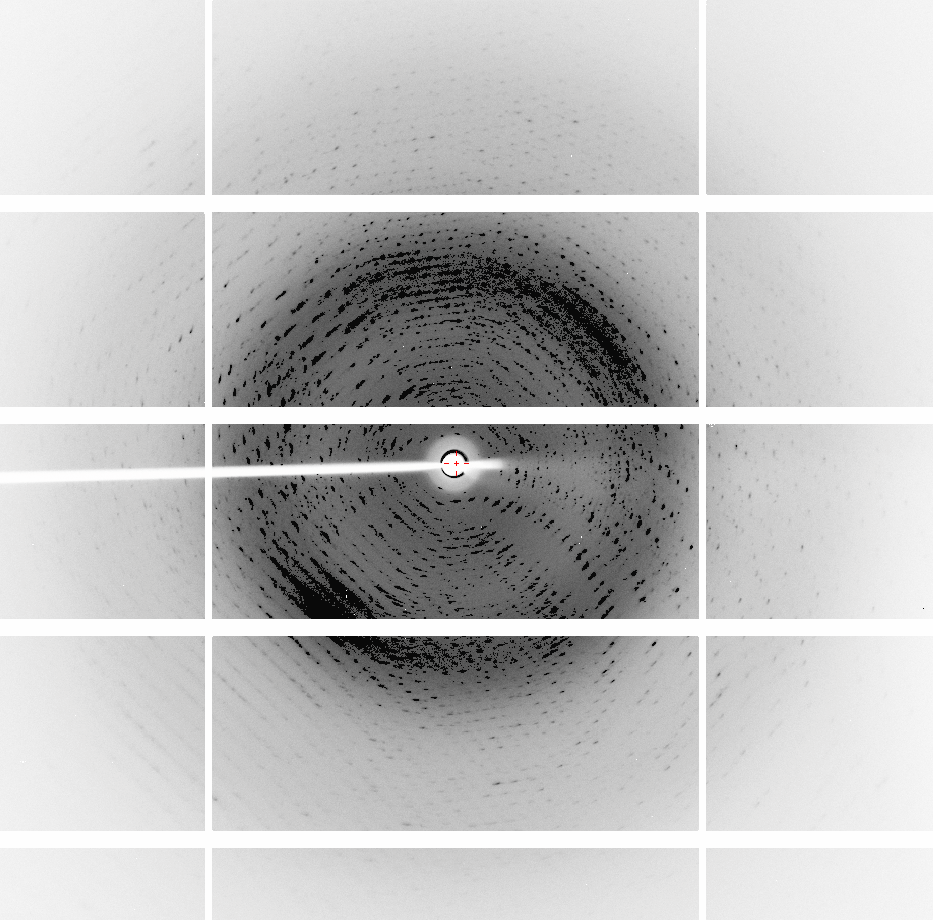
I have crystals of a 12.3 KDa protein that grow in hexagon-like patterns, link for the crystal image: http://fullonline.org/science/cryst01.jpg XDS, Phenix and Pointless always suggest that the data sets for these crystals belong to the space group P622. However, Phenix, Phaser and Pointless indicate that twinning is present. "Bad looking" diffraction images, diffracted to 1.6 A (collected 6 months ago): http://fullonline.org/science/dataset1_image37.png http://fullonline.org/science/dataset1_image7.png Best data set, diffracted to 2.01 A (collected a month ago): http://fullonline.org/science/dataset2_image51.png The second data set present better looking images, a better XDS ISa value (around 24) and diffracted to 2.2 A. The "bad looking" data set diffracted to 1.6 A, but I decided to stop working with it (XDS ISa around 10). There is a template with 60% identity, I used XDS to try to process the data in all trigonal/hexagonal space groups from P3 to P6(3)22, and spend a lot of time trying molecular replacement procedures in Phaser and Morda, and refining the candidate solutions. Used Zanuda too, trying to figure out the space group (and read as much as possible in this CCP4 list looking for similar cases). Unit cells and typical MR results: P1: 56.430 77.718 77.673 119.99 89.99 89.97 SOLU SET RFZ=9.5 TFZ=* PAK=0 LLG=91 RF++ TFZ=18.2 PAK=0 LLG=311 TFZ==18.4 (& TFZ==17.0) LLG+=(311 & 726) LLG=5751 TFZ==64.6 PAK=0 LLG=5751 TFZ==64.6 P3: 77.675 77.675 56.409 90.00 90.00 120.00 RFZ=5.5 TFZ=8.5 PAK=0 LLG=86 TFZ==9.4 LLG=2575 TFZ==46.5 P6: 77.534 77.534 92.986 90.00 90.00 120.00 RFZ=11.2 TFZ=16.2 PAK=2 LLG=437 TFZ==25.7 LLG=437 TFZ==25.7 P6(3): 77.675 77.675 56.409 90.00 90.00 120.00 RFZ=8.8 TFZ=11.5 PAK=0 LLG=66 TFZ==7.6 LLG=66 TFZ==7.6 P622: 77.660 77.660 56.400 90.00 90.00 120.00 RFZ=4.8 TFZ=5.4 PAK=1 LLG=22 TFZ==4.7 LLG=24 TFZ==4.8 In P1 (LLG=5751 TFZ==64.6) there are 12 molecules in the asymmetric unit, and in P3 (LLG=2575 TFZ==46.5) 4 molecules. Packing looks good, in P1 the ASU looks like two superposed hexagonal donuts formed by 6 molecules each. Refining in P1, without adding waters or TLS, yield r_work = 0.2964 and r_free = 0.3428, and it is hard to decrease these values. Refining in P3, I managed to get r_work = 0.2934 and r_free = 0.3399, but looks like it's not getting any better than this. Refining in P6 yields horrible r_free values (>0.50). Trying to refine MR solutions in any other other space groups yield Rfree 0.41 or more. If in the P3 space group I use the twin operator -k,-h,-l (estimated twin fraction of 0.490) suggested by Phenix Xtriage, the values miraculously go down to r_work = 0.1923 r_free = 0.2202, without waters (r_free without using a twin law = 0.3399). Adding 120 water molecules and doing some refining yields 0.1711 r_free = 0.1935 (the asymmetric unit contains 420 residues and the resolution is 2.01 A). I've been reading about twinning refinement and how it can drop the Rvalues, but from what I understood if I use it improperly I may compromise the refinement quality. I would like advice on: 1 - References that can teach how to look at protein diffraction images and understand what I am seeing. The basics like recognizing bad data and what usually leads to deformity in the spots (for example, elliptical or duplicated) would be of great help. 2 - Should I look for other space groups? What else could be tried? Is this a case where a twin law should be used in the refinement? If yes, what can I do to confirm the need for a twin law in the refinement? Thank you all in advance. Regards, Napo
{kind=link}
{kind=link}
{kind=link}
{kind=link}